torchdrug.data#
Data Structures#
Graph#
- class Graph(edge_list=None, edge_weight=None, num_node=None, num_relation=None, node_feature=None, edge_feature=None, graph_feature=None, **kwargs)[source]#
Basic container for sparse graphs.
To batch graphs with variadic sizes, use
data.Graph.pack. This will return a PackedGraph object with the following block diagonal adjacency matrix.\[\begin{split}\begin{bmatrix} A_1 & \cdots & 0 \\ \vdots & \ddots & \vdots \\ 0 & \cdots & A_n \end{bmatrix}\end{split}\]where \(A_i\) is the adjacency of \(i\)-th graph.
You may register dynamic attributes for each graph. The registered attributes will be automatically processed during packing.
Warning
This class doesn’t enforce any order on the edges.
Example:
>>> graph = data.Graph(torch.randint(10, (30, 2))) >>> with graph.node(): >>> graph.my_node_attr = torch.rand(10, 5, 5)
- Parameters
edge_list (array_like, optional) – list of edges of shape \((|E|, 2)\) or \((|E|, 3)\). Each tuple is (node_in, node_out) or (node_in, node_out, relation).
edge_weight (array_like, optional) – edge weights of shape \((|E|,)\)
num_node (int, optional) – number of nodes. By default, it will be inferred from the largest id in edge_list
num_relation (int, optional) – number of relations
node_feature (array_like, optional) – node features of shape \((|V|, ...)\)
edge_feature (array_like, optional) – edge features of shape \((|E|, ...)\)
graph_feature (array_like, optional) – graph feature of any shape
- packed_type#
alias of
torchdrug.data.graph.PackedGraph
- connected_components()[source]#
Split this graph into connected components.
- Returns
connected components, number of connected components per graph
- Return type
(PackedGraph, LongTensor)
- copy_(src)[source]#
Copy data from
srcintoselfand returnself.The
srcgraph must have the same set of attributes asself.
- cpu()[source]#
Return a copy of this graph in CPU memory.
This is a non-op if the graph is already in CPU memory.
- cuda(*args, **kwargs)[source]#
Return a copy of this graph in CUDA memory.
This is a non-op if the graph is already on the correct device.
- directed(order=None)[source]#
Mask the edges to create a directed graph. Edges that go from a node index to a larger or equal node index will be kept.
- Parameters
order (Tensor, optional) – topological order of the nodes
- edge_mask(index)[source]#
Return a masked graph based on the specified edges.
This function can also be used to re-order the edges.
- Parameters
index (array_like) – edge index
- Returns
Graph
- classmethod from_dense(adjacency, node_feature=None, edge_feature=None)[source]#
Create a sparse graph from a dense adjacency matrix. For zero entries in the adjacency matrix, their edge features will be ignored.
- Parameters
adjacency (array_like) – adjacency matrix of shape \((|V|, |V|)\) or \((|V|, |V|, |R|)\)
node_feature (array_like) – node features of shape \((|V|, ...)\)
edge_feature (array_like) – edge features of shape \((|V|, |V|, ...)\) or \((|V|, |V|, |R|, ...)\)
- get_edge(edge)[source]#
Get the weight of of an edge.
- Parameters
edge (array_like) – index of shape \((2,)\) or \((3,)\)
- Returns
weight of the edge
- Return type
Tensor
- line_graph()[source]#
Construct a line graph of this graph. The node feature of the line graph is inherited from the edge feature of the original graph.
In the line graph, each node corresponds to an edge in the original graph. For a pair of edges (a, b) and (b, c) that share the same intermediate node in the original graph, there is a directed edge (a, b) -> (b, c) in the line graph.
- Returns
Graph
- match(pattern)[source]#
Return all matched indexes for each pattern. Support patterns with
-1as the wildcard.- Parameters
pattern (array_like) – index of shape \((N, 2)\) or \((N, 3)\)
- Returns
matched indexes, number of matches per edge
- Return type
(LongTensor, LongTensor)
Examples:
>>> graph = data.Graph([[0, 1], [1, 0], [1, 2], [2, 1], [2, 0], [0, 2]]) >>> index, num_match = graph.match([[0, -1], [1, 2]]) >>> assert (index == torch.tensor([0, 5, 2])).all() >>> assert (num_match == torch.tensor([2, 1])).all()
- node_mask(index, compact=False)[source]#
Return a masked graph based on the specified nodes.
This function can also be used to re-order the nodes.
- Parameters
index (array_like) – node index
compact (bool, optional) – compact node ids or not
- Returns
Graph
Examples:
>>> graph = data.Graph.from_dense(torch.eye(3)) >>> assert graph.node_mask([1, 2]).adjacency.shape == (3, 3) >>> assert graph.node_mask([1, 2], compact=True).adjacency.shape == (2, 2)
- classmethod pack(graphs)[source]#
Pack a list of graphs into a PackedGraph object.
- Parameters
graphs (list of Graph) – list of graphs
- Returns
PackedGraph
- repeat(count)[source]#
Repeat this graph.
- Parameters
count (int) – number of repetitions
- Returns
PackedGraph
- split(node2graph)[source]#
Split a graph into multiple disconnected graphs.
- Parameters
node2graph (array_like) – ID of the graph each node belongs to
- Returns
PackedGraph
- subgraph(index)[source]#
Return a subgraph based on the specified nodes. Equivalent to
node_mask(index, compact=True).- Parameters
index (array_like) – node index
- Returns
Graph
See also
- undirected(add_inverse=False)[source]#
Flip all the edges to create an undirected graph.
For knowledge graphs, the flipped edges can either have the original relation or an inverse relation. The inverse relation for relation \(r\) is defined as \(|R| + r\).
- Parameters
add_inverse (bool, optional) – whether to use inverse relations for flipped edges
- visualize(title=None, save_file=None, figure_size=(3, 3), ax=None, layout='spring')[source]#
Visualize this graph with matplotlib.
- Parameters
title (str, optional) – title for this graph
save_file (str, optional) –
pngorpdffile to save visualization. If not provided, show the figure in window.figure_size (tuple of int, optional) – width and height of the figure
ax (matplotlib.axes.Axes, optional) – axis to plot the figure
layout (str, optional) – graph layout
See also
- property adjacency#
Adjacency matrix of this graph.
If
num_relationis specified, a sparse tensor of shape \((|V|, |V|, num\_relation)\) will be returned. Otherwise, a sparse tensor of shape \((|V|, |V|)\) will be returned.
- property batch_size#
Batch size.
- property degree_in#
Weighted number of edges containing each node as input.
Note this is the out-degree in graph theory.
- property degree_out#
Weighted number of edges containing each node as output.
Note this is the in-degree in graph theory.
- property device#
Device.
- property edge2graph#
Edge id to graph id mapping.
- property edge_list#
List of edges.
- property edge_weight#
Edge weights.
- property node2graph#
Node id to graph id mapping.
Molecule#
- class Molecule(edge_list=None, atom_type=None, bond_type=None, atom_feature=None, bond_feature=None, mol_feature=None, formal_charge=None, explicit_hs=None, chiral_tag=None, radical_electrons=None, atom_map=None, bond_stereo=None, stereo_atoms=None, node_position=None, **kwargs)[source]#
Molecules with predefined chemical features.
By nature, molecules are undirected graphs. Each bond is stored as two directed edges in this class.
Warning
This class doesn’t enforce any order on edges.
- Parameters
edge_list (array_like, optional) – list of edges of shape \((|E|, 3)\). Each tuple is (node_in, node_out, bond_type).
atom_type (array_like, optional) – atom types of shape \((|V|,)\)
bond_type (array_like, optional) – bond types of shape \((|E|,)\)
formal_charge (array_like, optional) – formal charges of shape \((|V|,)\)
explicit_hs (array_like, optional) – number of explicit hydrogens of shape \((|V|,)\)
chiral_tag (array_like, optional) – chirality tags of shape \((|V|,)\)
radical_electrons (array_like, optional) – number of radical electrons of shape \((|V|,)\)
atom_map (array_likeb optional) – atom mappings of shape \((|V|,)\)
bond_stereo (array_like, optional) – bond stereochem of shape \((|E|,)\)
stereo_atoms (array_like, optional) – ids of stereo atoms of shape \((|E|,)\)
- packed_type#
- edge_mask(index)[source]#
Return a masked graph based on the specified edges.
This function can also be used to re-order the edges.
- Parameters
index (array_like) – edge index
- Returns
Graph
- classmethod from_molecule(cls, mol, atom_feature='default', bond_feature='default', mol_feature=None, with_hydrogen=False, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Create a molecule from an RDKit object.
- Parameters
mol (rdchem.Mol) – molecule
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
mol_feature (str or list of str, optional) – molecule features to extract
with_hydrogen (bool, optional) – store hydrogens in the molecule graph. By default, hydrogens are dropped
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- classmethod from_smiles(cls, smiles, atom_feature='default', bond_feature='default', mol_feature=None, with_hydrogen=False, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Create a molecule from a SMILES string.
- Parameters
smiles (str) – SMILES string
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
mol_feature (str or list of str, optional) – molecule features to extract
with_hydrogen (bool, optional) – store hydrogens in the molecule graph. By default, hydrogens are dropped
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- ion_to_molecule()[source]#
Convert ions to molecules by adjusting hydrogens and electrons.
Note [N+] will not be converted.
- node_mask(index, compact=False)[source]#
Return a masked graph based on the specified nodes.
This function can also be used to re-order the nodes.
- Parameters
index (array_like) – node index
compact (bool, optional) – compact node ids or not
- Returns
Graph
Examples:
>>> graph = data.Graph.from_dense(torch.eye(3)) >>> assert graph.node_mask([1, 2]).adjacency.shape == (3, 3) >>> assert graph.node_mask([1, 2], compact=True).adjacency.shape == (2, 2)
- to_molecule(ignore_error=False)[source]#
Return an RDKit object of this molecule.
- Parameters
ignore_error (bool, optional) – if true, return
Nonefor illegal molecules. Otherwise, raise an exception.- Returns
rdchem.Mol
- to_scaffold(chirality=False)[source]#
Return a scaffold SMILES string of this molecule.
- Parameters
chirality (bool, optional) – consider chirality in the scaffold or not
- Returns
str
- to_smiles(isomeric=True, atom_map=True, canonical=False)[source]#
Return a SMILES string of this molecule.
- Parameters
isomeric (bool, optional) – keep isomeric information or not
atom_map (bool, optional) – keep atom mapping or not
canonical (bool, optional) – if true, return the canonical form of smiles
- Returns
str
- undirected(add_inverse=False)[source]#
Flip all the edges to create an undirected graph.
For knowledge graphs, the flipped edges can either have the original relation or an inverse relation. The inverse relation for relation \(r\) is defined as \(|R| + r\).
- Parameters
add_inverse (bool, optional) – whether to use inverse relations for flipped edges
- visualize(title=None, save_file=None, figure_size=(3, 3), ax=None, atom_map=False)[source]#
Visualize this molecule with matplotlib.
- Parameters
title (str, optional) – title for this molecule
save_file (str, optional) –
pngorpdffile to save visualization. If not provided, show the figure in window.figure_size (tuple of int, optional) – width and height of the figure
ax (matplotlib.axes.Axes, optional) – axis to plot the figure
atom_map (bool, optional) – visualize atom mapping or not
- property atom2graph#
Node id to graph id mapping.
- property bond2graph#
Edge id to graph id mapping.
- property is_valid#
A coarse implementation of valence check.
Protein#
- class Protein(edge_list=None, atom_type=None, bond_type=None, residue_type=None, view=None, atom_name=None, atom2residue=None, residue_feature=None, is_hetero_atom=None, occupancy=None, b_factor=None, residue_number=None, insertion_code=None, chain_id=None, **kwargs)[source]#
Proteins with predefined chemical features. Support both residue-level and atom-level operations and ensure consistency between two views.
Warning
The order of residues must be the same as the protein sequence. However, this class doesn’t enforce any order on nodes or edges. Nodes may have a different order with residues.
- Parameters
edge_list (array_like, optional) – list of edges of shape \((|E|, 3)\). Each tuple is (node_in, node_out, bond_type).
atom_type (array_like, optional) – atom types of shape \((|V|,)\)
bond_type (array_like, optional) – bond types of shape \((|E|,)\)
residue_type (array_like, optional) – residue types of shape \((|V_{res}|,)\)
view (str, optional) – default view for this protein. Can be
atomorresidue.atom_name (array_like, optional) – atom names in a residue of shape \((|V|,)\)
atom2residue (array_like, optional) – atom id to residue id mapping of shape \((|V|,)\)
residue_feature (array_like, optional) – residue features of shape \((|V_{res}|, ...)\)
is_hetero_atom (array_like, optional) – hetero atom indicators of shape \((|V|,)\)
occupancy (array_like, optional) – occupancy of shape \((|V|,)\)
b_factor (array_like, optional) – temperature factors of shape \((|V|,)\)
residue_number (array_like, optional) – residue numbers of shape \((|V_{res}|,)\)
insertion_code (array_like, optional) – insertion codes of shape \((|V_{res}|,)\)
chain_id (array_like, optional) – chain ids of shape \((|V_{res}|,)\)
- packed_type#
alias of
torchdrug.data.protein.PackedProtein
- classmethod from_molecule(cls, mol, atom_feature='default', bond_feature='default', residue_feature='default', mol_feature=None, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Create a protein from an RDKit object.
- Parameters
mol (rdchem.Mol) – molecule
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
residue_feature (str, list of str, optional) – residue features to extract
mol_feature (str or list of str, optional) – molecule features to extract
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- classmethod from_pdb(cls, pdb_file, atom_feature='default', bond_feature='default', residue_feature='default', mol_feature=None, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Create a protein from a PDB file.
- Parameters
pdb_file (str) – file name
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
residue_feature (str, list of str, optional) – residue features to extract
mol_feature (str or list of str, optional) – molecule features to extract
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- classmethod from_sequence(cls, sequence, atom_feature='default', bond_feature='default', residue_feature='default', mol_feature=None, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Create a protein from a sequence.
Note
It takes considerable time to construct proteins with a large number of atoms and bonds. If you only need residue information, you may speed up the construction by setting
atom_featureandbond_featuretoNone.- Parameters
sequence (str) – protein sequence
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
residue_feature (str, list of str, optional) – residue features to extract
mol_feature (str or list of str, optional) – molecule features to extract
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- classmethod pack(graphs)[source]#
Pack a list of graphs into a PackedGraph object.
- Parameters
graphs (list of Graph) – list of graphs
- Returns
PackedGraph
- repeat(count)[source]#
Repeat this graph.
- Parameters
count (int) – number of repetitions
- Returns
PackedGraph
- residue_mask(index, compact=False)[source]#
Return a masked protein based on the specified residues.
Note the compact option is applied to both residue and atom ids.
- Parameters
index (array_like) – residue index
compact (bool, optional) – compact residue ids or not
- Returns
Protein
- split(node2graph)[source]#
Split a graph into multiple disconnected graphs.
- Parameters
node2graph (array_like) – ID of the graph each node belongs to
- Returns
PackedGraph
- subresidue(index)[source]#
Return a subgraph based on the specified residues. Equivalent to
residue_mask(index, compact=True).- Parameters
index (array_like) – residue index
- Returns
Protein
See also
- to_molecule(ignore_error=False)[source]#
Return an RDKit object of this protein.
- Parameters
ignore_error (bool, optional) – if true, return
Nonefor illegal molecules. Otherwise, raise an exception.- Returns
rdchem.Mol
- property connected_component_id#
Connected component id of each residue.
- property residue2graph#
Residue id to graph id mapping.
PackedGraph#
- class PackedGraph(edge_list=None, edge_weight=None, num_nodes=None, num_edges=None, num_relation=None, offsets=None, **kwargs)[source]#
Container for sparse graphs with variadic sizes.
To create a PackedGraph from Graph objects
>>> batch = data.Graph.pack(graphs)
To retrieve Graph objects from a PackedGraph
>>> graphs = batch.unpack()
Warning
Edges of the same graph are guaranteed to be consecutive in the edge list. However, this class doesn’t enforce any order on the edges.
- Parameters
edge_list (array_like, optional) – list of edges of shape \((|E|, 2)\) or \((|E|, 3)\). Each tuple is (node_in, node_out) or (node_in, node_out, relation).
edge_weight (array_like, optional) – edge weights of shape \((|E|,)\)
num_nodes (array_like, optional) – number of nodes in each graph By default, it will be inferred from the largest id in edge_list
num_edges (array_like, optional) – number of edges in each graph
num_relation (int, optional) – number of relations
node_feature (array_like, optional) – node features of shape \((|V|, ...)\)
edge_feature (array_like, optional) – edge features of shape \((|E|, ...)\)
offsets (array_like, optional) – node id offsets of shape \((|E|,)\). If not provided, nodes in edge_list should be relative index, i.e., the index in each graph. If provided, nodes in edge_list should be absolute index, i.e., the index in the packed graph.
- unpacked_type#
alias of
torchdrug.data.graph.Graph
- cpu()[source]#
Return a copy of this packed graph in CPU memory.
This is a non-op if the graph is already in CPU memory.
- cuda(*args, **kwargs)[source]#
Return a copy of this packed graph in CUDA memory.
This is a non-op if the graph is already on the correct device.
- edge_mask(index)[source]#
Return a masked packed graph based on the specified edges.
- Parameters
index (array_like) – edge index
- Returns
PackedGraph
- full()[source]#
Return a pack of fully connected graphs.
This is useful for computing node-pair-wise features. The computation can be implemented as message passing over a fully connected graph.
- Returns
PackedGraph
- get_item(index)[source]#
Get the i-th graph from this packed graph.
- Parameters
index (int) – graph index
- Returns
Graph
- graph_mask(index, compact=False)[source]#
Return a masked packed graph based on the specified graphs.
This function can also be used to re-order the graphs.
- Parameters
index (array_like) – graph index
compact (bool, optional) – compact graph ids or not
- Returns
PackedGraph
- line_graph()[source]#
Construct a packed line graph of this packed graph. The node features of the line graphs are inherited from the edge features of the original graphs.
In the line graph, each node corresponds to an edge in the original graph. For a pair of edges (a, b) and (b, c) that share the same intermediate node in the original graph, there is a directed edge (a, b) -> (b, c) in the line graph.
- Returns
PackedGraph
- merge(graph2graph)[source]#
Merge multiple graphs into a single graph.
- Parameters
graph2graph (array_like) – ID of the new graph each graph belongs to
- node_mask(index, compact=False)[source]#
Return a masked packed graph based on the specified nodes.
Note the compact option is only applied to node ids but not graph ids. To generate compact graph ids, use
subbatch().- Parameters
index (array_like) – node index
compact (bool, optional) – compact node ids or not
- Returns
PackedGraph
- repeat(count)[source]#
Repeat this packed graph. This function behaves similarly to torch.Tensor.repeat.
- Parameters
count (int) – number of repetitions
- Returns
PackedGraph
- repeat_interleave(repeats)[source]#
Repeat this packed graph. This function behaves similarly to torch.repeat_interleave.
- Parameters
repeats (Tensor or int) – number of repetitions for each graph
- Returns
PackedGraph
- subbatch(index)[source]#
Return a subbatch based on the specified graphs. Equivalent to
graph_mask(index, compact=True).- Parameters
index (array_like) – graph index
- Returns
PackedGraph
See also
- undirected(add_inverse=False)[source]#
Flip all the edges to create undirected graphs.
For knowledge graphs, the flipped edges can either have the original relation or an inverse relation. The inverse relation for relation \(r\) is defined as \(|R| + r\).
- Parameters
add_inverse (bool, optional) – whether to use inverse relations for flipped edges
- unpack_data(data, type='auto')[source]#
Unpack node or edge data according to the packed graph.
- Parameters
data (Tensor) – data to unpack
type (str, optional) – data type. Can be
auto,node, oredge.
- Returns
list of Tensor
- visualize(titles=None, save_file=None, figure_size=(3, 3), layout='spring', num_row=None, num_col=None)[source]#
Visualize the packed graphs with matplotlib.
- Parameters
titles (list of str, optional) – title for each graph. Default is the ID of each graph.
save_file (str, optional) –
pngorpdffile to save visualization. If not provided, show the figure in window.figure_size (tuple of int, optional) – width and height of the figure
layout (str, optional) – graph layout
num_row (int, optional) – number of rows in the figure
num_col (int, optional) – number of columns in the figure
See also
- property batch_size#
Batch size.
- property edge2graph#
Edge id to graph id mapping.
- property node2graph#
Node id to graph id mapping.
PackedMolecule#
- class PackedMolecule(edge_list=None, atom_type=None, bond_type=None, num_nodes=None, num_edges=None, offsets=None, **kwargs)[source]#
Container for molecules with variadic sizes.
Warning
Edges of the same molecule are guaranteed to be consecutive in the edge list. However, this class doesn’t enforce any order on the edges.
- Parameters
edge_list (array_like, optional) – list of edges of shape \((|E|, 3)\). Each tuple is (node_in, node_out, bond_type).
atom_type (array_like, optional) – atom types of shape \((|V|,)\)
bond_type (array_like, optional) – bond types of shape \((|E|,)\)
num_nodes (array_like, optional) – number of nodes in each graph By default, it will be inferred from the largest id in edge_list
num_edges (array_like, optional) – number of edges in each graph
offsets (array_like, optional) – node id offsets of shape \((|E|,)\). If not provided, nodes in edge_list should be relative index, i.e., the index in each graph. If provided, nodes in edge_list should be absolute index, i.e., the index in the packed graph.
- unpacked_type#
alias of
torchdrug.data.molecule.Molecule
- edge_mask(index)[source]#
Return a masked packed graph based on the specified edges.
- Parameters
index (array_like) – edge index
- Returns
PackedGraph
- classmethod from_molecule(cls, mols, atom_feature='default', bond_feature='default', mol_feature=None, with_hydrogen=False, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Create a packed molecule from a list of RDKit objects.
- Parameters
mols (list of rdchem.Mol) – molecules
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
mol_feature (str or list of str, optional) – molecule features to extract
with_hydrogen (bool, optional) – store hydrogens in the molecule graph. By default, hydrogens are dropped
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- classmethod from_smiles(cls, smiles_list, atom_feature='default', bond_feature='default', mol_feature=None, with_hydrogen=False, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Create a packed molecule from a list of SMILES strings.
- Parameters
smiles_list (str) – list of SMILES strings
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
mol_feature (str or list of str, optional) – molecule features to extract
with_hydrogen (bool, optional) – store hydrogens in the molecule graph. By default, hydrogens are dropped
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- ion_to_molecule()[source]#
Convert ions to molecules by adjusting hydrogens and electrons.
Note [N+] will not be converted.
- node_mask(index, compact=False)[source]#
Return a masked packed graph based on the specified nodes.
Note the compact option is only applied to node ids but not graph ids. To generate compact graph ids, use
subbatch().- Parameters
index (array_like) – node index
compact (bool, optional) – compact node ids or not
- Returns
PackedGraph
- to_molecule(ignore_error=False)[source]#
Return a list of RDKit objects.
- Parameters
ignore_error (bool, optional) – if true, return
Nonefor illegal molecules. Otherwise, raise an exception.- Returns
list of rdchem.Mol
- to_smiles(isomeric=True, atom_map=True, canonical=False)[source]#
Return a list of SMILES strings.
- Parameters
isomeric (bool, optional) – keep isomeric information or not
atom_map (bool, optional) – keep atom mapping or not
canonical (bool, optional) – if true, return the canonical form of smiles
- Returns
list of str
- undirected(add_inverse=False)[source]#
Flip all the edges to create undirected graphs.
For knowledge graphs, the flipped edges can either have the original relation or an inverse relation. The inverse relation for relation \(r\) is defined as \(|R| + r\).
- Parameters
add_inverse (bool, optional) – whether to use inverse relations for flipped edges
- visualize(titles=None, save_file=None, figure_size=(3, 3), num_row=None, num_col=None, atom_map=False)[source]#
Visualize the packed molecules with matplotlib.
- Parameters
titles (list of str, optional) – title for each molecule. Default is the ID of each molecule.
save_file (str, optional) –
pngorpdffile to save visualization. If not provided, show the figure in window.figure_size (tuple of int, optional) – width and height of the figure
num_row (int, optional) – number of rows in the figure
num_col (int, optional) – number of columns in the figure
atom_map (bool, optional) – visualize atom mapping or not
- property atom2graph#
Node id to graph id mapping.
- property bond2graph#
Edge id to graph id mapping.
- property is_valid#
A coarse implementation of valence check.
PackedProtein#
- class PackedProtein(edge_list=None, atom_type=None, bond_type=None, residue_type=None, view=None, num_nodes=None, num_edges=None, num_residues=None, offsets=None, **kwargs)[source]#
Container for proteins with variadic sizes. Support both residue-level and atom-level operations and ensure consistency between two views.
Warning
Edges of the same graph are guaranteed to be consecutive in the edge list. The order of residues must be the same as the protein sequence. However, this class doesn’t enforce any order on nodes or edges. Nodes may have a different order with residues.
- Parameters
edge_list (array_like, optional) – list of edges of shape \((|E|, 3)\). Each tuple is (node_in, node_out, bond_type).
atom_type (array_like, optional) – atom types of shape \((|V|,)\)
bond_type (array_like, optional) – bond types of shape \((|E|,)\)
residue_type (array_like, optional) – residue types of shape \((|V_{res}|,)\)
view (str, optional) – default view for this protein. Can be
atomorresidue.num_nodes (array_like, optional) – number of nodes in each graph By default, it will be inferred from the largest id in edge_list
num_edges (array_like, optional) – number of edges in each graph
num_residues (array_like, optional) – number of residues in each graph
offsets (array_like, optional) – node id offsets of shape \((|E|,)\). If not provided, nodes in edge_list should be relative index, i.e., the index in each graph. If provided, nodes in edge_list should be absolute index, i.e., the index in the packed graph.
- unpacked_type#
alias of
torchdrug.data.protein.Protein
- cpu()[source]#
Return a copy of this packed graph in CPU memory.
This is a non-op if the graph is already in CPU memory.
- cuda(*args, **kwargs)[source]#
Return a copy of this packed graph in CUDA memory.
This is a non-op if the graph is already on the correct device.
- edge_mask(index)[source]#
Return a masked packed graph based on the specified edges.
- Parameters
index (array_like) – edge index
- Returns
PackedGraph
- classmethod from_molecule(cls, mols, atom_feature='default', bond_feature='default', residue_feature='default', mol_feature=None, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Create a packed protein from a list of RDKit objects.
- Parameters
mols (list of rdchem.Mol) – molecules
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
residue_feature (str or list of str, optional) – residue features to extract
mol_feature (str or list of str, optional) – molecule features to extract
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- classmethod from_pdb(cls, pdb_files, atom_feature='default', bond_feature='default', residue_feature='default', mol_feature=None, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Create a protein from a list of PDB files.
- Parameters
pdb_files (str) – list of file names
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
residue_feature (str, list of str, optional) – residue features to extract
mol_feature (str or list of str, optional) – molecule features to extract
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- classmethod from_sequence(cls, sequences, atom_feature='default', bond_feature='default', residue_feature='default', mol_feature=None, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Create a packed protein from a list of sequences.
Note
It takes considerable time to construct proteins with a large number of atoms and bonds. If you only need residue information, you may speed up the construction by setting
atom_featureandbond_featuretoNone.- Parameters
sequences (str) – list of protein sequences
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
residue_feature (str or list of str, optional) – residue features to extract
mol_feature (str or list of str, optional) – molecule features to extract
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- get_item(index)[source]#
Get the i-th graph from this packed graph.
- Parameters
index (int) – graph index
- Returns
Graph
- graph_mask(index, compact=False)[source]#
Return a masked packed graph based on the specified graphs.
This function can also be used to re-order the graphs.
- Parameters
index (array_like) – graph index
compact (bool, optional) – compact graph ids or not
- Returns
PackedGraph
- merge(graph2graph)[source]#
Merge multiple graphs into a single graph.
- Parameters
graph2graph (array_like) – ID of the new graph each graph belongs to
- node_mask(index, compact=True)[source]#
Return a masked packed graph based on the specified nodes.
Note the compact option is only applied to node ids but not graph ids. To generate compact graph ids, use
subbatch().- Parameters
index (array_like) – node index
compact (bool, optional) – compact node ids or not
- Returns
PackedGraph
- repeat(count)[source]#
Repeat this packed graph. This function behaves similarly to torch.Tensor.repeat.
- Parameters
count (int) – number of repetitions
- Returns
PackedGraph
- repeat_interleave(repeats)[source]#
Repeat this packed graph. This function behaves similarly to torch.repeat_interleave.
- Parameters
repeats (Tensor or int) – number of repetitions for each graph
- Returns
PackedGraph
- residue_mask(index, compact=False)[source]#
Return a masked packed protein based on the specified residues.
Note the compact option is applied to both residue and atom ids, but not graph ids.
- Parameters
index (array_like) – residue index
compact (bool, optional) – compact residue ids or not
- Returns
PackedProtein
- to_molecule(ignore_error=False)[source]#
Return a list of RDKit objects.
- Parameters
ignore_error (bool, optional) – if true, return
Nonefor illegal molecules. Otherwise, raise an exception.- Returns
list of rdchem.Mol
- to_pdb(pdb_files)[source]#
Write this packed protein to several pdb files.
- Parameters
pdb_files (list of str) – list of file names
- undirected(add_inverse=True)[source]#
Flip all the edges to create undirected graphs.
For knowledge graphs, the flipped edges can either have the original relation or an inverse relation. The inverse relation for relation \(r\) is defined as \(|R| + r\).
- Parameters
add_inverse (bool, optional) – whether to use inverse relations for flipped edges
- property connected_component_id#
Connected component id of each residue.
- property residue2graph#
Residue id to graph id mapping.
Dictionary#
- class Dictionary(keys, values, hash=None)[source]#
Dictionary for mapping keys to values.
This class has the same behavior as the built-in dict, except it operates on tensors and support batching.
Example:
>>> keys = torch.tensor([[0, 0], [1, 1], [2, 2]]) >>> values = torch.tensor([[0, 1], [1, 2], [2, 3]]) >>> d = data.Dictionary(keys, values) >>> assert (d[[[0, 0], [2, 2]]] == values[[0, 2]]).all() >>> assert (d.has_key([[0, 1], [1, 2]]) == torch.tensor([False, False])).all()
- Parameters
keys (LongTensor) – keys of shape \((N,)\) or \((N, D)\)
values (Tensor) – values of shape \((N, ...)\)
hash (PerfectHash, optional) – hash function for keys
- cpu()[source]#
Return a copy of this dictionary in CPU memory.
This is a non-op if the dictionary is already in CPU memory.
- cuda(*args, **kwargs)[source]#
Return a copy of this dictionary in CUDA memory.
This is a non-op if the dictionary is already in CUDA memory.
- get(keys, default=None)[source]#
Return the value for each key if the key is in the dictionary, otherwise the default value is returned.
- Parameters
keys (LongTensor) – keys of arbitrary shape
default (int or Tensor, optional) – default return value. By default, 0 is used.
- property device#
Device.
Datasets#
KnowledgeGraphDataset#
- class KnowledgeGraphDataset(*args, **kwds)[source]#
Knowledge graph dataset.
The whole dataset contains one knowledge graph.
- load_triplet(triplets, entity_vocab=None, relation_vocab=None, inv_entity_vocab=None, inv_relation_vocab=None)[source]#
Load the dataset from triplets. The mapping between indexes and tokens is specified through either vocabularies or inverse vocabularies.
- Parameters
triplets (array_like) – triplets of shape \((n, 3)\)
entity_vocab (dict of str, optional) – maps entity indexes to tokens
relation_vocab (dict of str, optional) – maps relation indexes to tokens
inv_entity_vocab (dict of str, optional) – maps tokens to entity indexes
inv_relation_vocab (dict of str, optional) – maps tokens to relation indexes
- load_tsv(tsv_file, verbose=0)[source]#
Load the dataset from a tsv file.
- Parameters
tsv_file (str) – file name
verbose (int, optional) – output verbose level
- load_tsvs(tsv_files, verbose=0)[source]#
Load the dataset from multiple tsv files.
- Parameters
tsv_files (list of str) – list of file names
verbose (int, optional) – output verbose level
- property num_entity#
Number of entities.
- property num_relation#
Number of relations.
- property num_triplet#
Number of triplets.
MoleculeDataset#
- class MoleculeDataset(*args, **kwds)[source]#
Molecule dataset.
Each sample contains a molecule graph, and any number of prediction targets.
- load_csv(csv_file, smiles_field='smiles', target_fields=None, verbose=0, transform=None, lazy=False, atom_feature='default', bond_feature='default', mol_feature=None, with_hydrogen=False, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Load the dataset from a csv file.
- Parameters
csv_file (str) – file name
smiles_field (str, optional) – name of the SMILES column in the table. Use
Noneif there is no SMILES column.target_fields (list of str, optional) – name of target columns in the table. Default is all columns other than the SMILES column.
verbose (int, optional) – output verbose level
transform (Callable, optional) – data transformation function
lazy (bool, optional) – if lazy mode is used, the molecules are processed in the dataloader. This may slow down the data loading process, but save a lot of CPU memory and dataset loading time.
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
mol_feature (str or list of str, optional) – molecule features to extract
with_hydrogen (bool, optional) – store hydrogens in the molecule graph. By default, hydrogens are dropped
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- load_pickle(pkl_file, verbose=0)[source]#
Load the dataset from a pickle file.
- Parameters
pkl_file (str) – file name
verbose (int, optional) – output verbose level
- load_smiles(smiles_list, targets, transform=None, lazy=False, verbose=0, atom_feature='default', bond_feature='default', mol_feature=None, with_hydrogen=False, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Load the dataset from SMILES and targets.
- Parameters
smiles_list (list of str) – SMILES strings
targets (dict of list) – prediction targets
transform (Callable, optional) – data transformation function
lazy (bool, optional) – if lazy mode is used, the molecules are processed in the dataloader. This may slow down the data loading process, but save a lot of CPU memory and dataset loading time.
verbose (int, optional) – output verbose level
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
mol_feature (str or list of str, optional) – molecule features to extract
with_hydrogen (bool, optional) – store hydrogens in the molecule graph. By default, hydrogens are dropped
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- save_pickle(pkl_file, verbose=0)[source]#
Save the dataset to a pickle file.
- Parameters
pkl_file (str) – file name
verbose (int, optional) – output verbose level
- property atom_types#
All atom types.
- property bond_types#
All bond types.
- property edge_feature_dim#
Dimension of edge features.
- property node_feature_dim#
Dimension of node features.
- property num_atom_type#
Number of different atom types.
- property num_bond_type#
Number of different bond types.
- property tasks#
List of tasks.
ProteinDataset#
- class ProteinDataset(*args, **kwds)[source]#
Protein dataset.
Each sample contains a protein graph, and any number of prediction targets.
- load_fasta(fasta_file, verbose=0, attributes=None, transform=None, lazy=False, atom_feature='default', bond_feature='default', residue_feature='default', mol_feature=None, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Load the dataset from a fasta file.
- Parameters
fasta_file (str) – file name
verbose (int, optional) – output verbose level
attributes (dict of list) – protein-level attributes
transform (Callable, optional) – protein sequence transformation function
lazy (bool, optional) – if lazy mode is used, the proteins are processed in the dataloader. This may slow down the data loading process, but save a lot of CPU memory and dataset loading time.
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
residue_feature (str, list of str, optional) – residue features to extract
mol_feature (str or list of str, optional) – molecule features to extract
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- load_lmdbs(lmdb_files, sequence_field='primary', target_fields=None, number_field='num_examples', transform=None, lazy=False, verbose=0, attributes=None, atom_feature='default', bond_feature='default', residue_feature='default', mol_feature=None, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Load the dataset from lmdb files.
- Parameters
lmdb_files (list of str) – list of lmdb files
sequence_field (str, optional) – name of the field of protein sequence in lmdb files
target_fields (list of str, optional) – name of target fields in lmdb files
number_field (str, optional) – name of the field of sample count in lmdb files
transform (Callable, optional) – protein sequence transformation function
lazy (bool, optional) – if lazy mode is used, the proteins are processed in the dataloader. This may slow down the data loading process, but save a lot of CPU memory and dataset loading time.
verbose (int, optional) – output verbose level
attributes (dict of list) – protein-level attributes
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
residue_feature (str, list of str, optional) – residue features to extract
mol_feature (str or list of str, optional) – molecule features to extract
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- load_pdbs(pdb_files, transform=None, lazy=False, verbose=0, atom_feature='default', bond_feature='default', residue_feature='default', mol_feature=None, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Load the dataset from pdb files.
- Parameters
pdb_files (list of str) – pdb file names
transform (Callable, optional) – protein sequence transformation function
lazy (bool, optional) – if lazy mode is used, the proteins are processed in the dataloader. This may slow down the data loading process, but save a lot of CPU memory and dataset loading time.
verbose (int, optional) – output verbose level
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
residue_feature (str, list of str, optional) – residue features to extract
mol_feature (str or list of str, optional) – molecule features to extract
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- load_pickle(pkl_file, transform=None, lazy=False, verbose=0, atom_feature='default', bond_feature='default', residue_feature='default', mol_feature=None, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Load the dataset from a pickle file.
- Parameters
pkl_file (str) – file name
transform (Callable, optional) – protein sequence transformation function
lazy (bool, optional) – if lazy mode is used, the proteins are processed in the dataloader. This may slow down the data loading process, but save a lot of CPU memory and dataset loading time.
verbose (int, optional) – output verbose level
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
residue_feature (str, list of str, optional) – residue features to extract
mol_feature (str or list of str, optional) – molecule features to extract
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- load_sequence(sequences, targets, attributes=None, transform=None, lazy=False, verbose=0, atom_feature='default', bond_feature='default', residue_feature='default', mol_feature=None, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Load the dataset from protein sequences and targets.
- Parameters
sequences (list of str) – protein sequence strings
targets (dict of list) – prediction targets
attributes (dict of list) – protein-level attributes
transform (Callable, optional) – protein sequence transformation function
lazy (bool, optional) – if lazy mode is used, the proteins are processed in the dataloader. This may slow down the data loading process, but save a lot of CPU memory and dataset loading time.
verbose (int, optional) – output verbose level
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
residue_feature (str, list of str, optional) – residue features to extract
mol_feature (str or list of str, optional) – molecule features to extract
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- save_pickle(pkl_file, verbose=0)[source]#
Save the dataset to a pickle file.
- Parameters
pkl_file (str) – file name
verbose (int, optional) – output verbose level
- property residue_feature_dim#
Dimension of residue features.
ProteinPairDataset#
- class ProteinPairDataset(*args, **kwds)[source]#
Protein pair dataset.
Each sample contains two protein graphs, and any number of prediction targets.
- load_lmdbs(lmdb_files, sequence_field='primary', target_fields=None, number_field='num_examples', transform=None, lazy=False, verbose=0, attributes=None, atom_feature='default', bond_feature='default', residue_feature='default', mol_feature=None, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Load the dataset from lmdb files.
- Parameters
lmdb_files (list of str) – file names
sequence_field (str or list of str, optional) – names of the fields of protein sequence in lmdb files
target_fields (list of str, optional) – name of target fields in lmdb files
number_field (str, optional) – name of the field of sample count in lmdb files
transform (Callable, optional) – protein sequence transformation function
lazy (bool, optional) – if lazy mode is used, the protein pairs are processed in the dataloader. This may slow down the data loading process, but save a lot of CPU memory and dataset loading time.
verbose (int, optional) – output verbose level
attributes (dict of list) – protein-level attributes
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
residue_feature (str, list of str, optional) – residue features to extract
mol_feature (str or list of str, optional) – molecule features to extract
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- load_sequence(sequences, targets, attributes=None, transform=None, lazy=False, verbose=0, atom_feature='default', bond_feature='default', residue_feature='default', mol_feature=None, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Load the dataset from protein sequences and targets.
- Parameters
sequences (list of list of str) – protein sequence string pairs
targets (dict of list) – prediction targets
attributes (dict of list) – protein-level attributes
transform (Callable, optional) – protein sequence transformation function
lazy (bool, optional) – if lazy mode is used, the protein pairs are processed in the dataloader. This may slow down the data loading process, but save a lot of CPU memory and dataset loading time.
verbose (int, optional) – output verbose level
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
residue_feature (str, list of str, optional) – residue features to extract
mol_feature (str or list of str, optional) – molecule features to extract
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- property node_feature_dim#
Dimension of node features.
- property residue_feature_dim#
Dimension of residue features.
ProteinLigandDataset#
- class ProteinLigandDataset(*args, **kwds)[source]#
Protein-ligand dataset.
Each sample contains a protein graph and a molecule graph, and any number of prediction targets.
- load_lmdbs(lmdb_files, sequence_field='target', smiles_field='drug', target_fields=None, number_field='num_examples', transform=None, lazy=False, verbose=0, attributes=None, atom_feature='default', bond_feature='default', residue_feature='default', mol_feature=None, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Load the dataset from lmdb files.
- Parameters
lmdb_files (list of str) – file names
sequence_field (str, optional) – name of the field of protein sequence in lmdb files
smiles_field (str, optional) – name of the field of ligand SMILES string in lmdb files
target_fields (list of str, optional) – name of target fields in lmdb files
number_field (str, optional) – name of the field of sample count in lmdb files
transform (Callable, optional) – protein sequence transformation function
lazy (bool, optional) – if lazy mode is used, the protein-ligand pairs are processed in the dataloader. This may slow down the data loading process, but save a lot of CPU memory and dataset loading time.
verbose (int, optional) – output verbose level
attributes (dict of list) – protein-level attributes
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
residue_feature (str, list of str, optional) – residue features to extract
mol_feature (str or list of str, optional) – molecule features to extract
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- load_sequence(sequences, smiles, targets, num_samples, attributes=None, transform=None, lazy=False, verbose=0, atom_feature='default', bond_feature='default', residue_feature='default', mol_feature=None, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Load the dataset from protein sequences, ligand SMILES strings and targets.
- Parameters
sequences (list of str) – protein sequence strings
smiles (list of str) – ligand SMILES strings
targets (dict of list) – prediction targets
num_samples (list of int) – numbers of protein-ligand pairs in all splits
attributes (dict of list) – protein-level attributes
transform (Callable, optional) – protein sequence transformation function
lazy (bool, optional) – if lazy mode is used, the protein-ligand pairs are processed in the dataloader. This may slow down the data loading process, but save a lot of CPU memory and dataset loading time.
verbose (int, optional) – output verbose level
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
residue_feature (str, list of str, optional) – residue features to extract
mol_feature (str or list of str, optional) – molecule features to extract
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- property ligand_node_feature_dim#
Dimension of node features for ligands.
- property protein_node_feature_dim#
Dimension of node features for proteins.
- property residue_feature_dim#
Dimension of residue features for proteins.
NodeClassificationDataset#
- class NodeClassificationDataset(*args, **kwds)[source]#
Node classification dataset.
The whole dataset contains one graph, where each node has its own node feature and label.
- load_tsv(node_file, edge_file, verbose=0)[source]#
Load the edge list from a tsv file.
- Parameters
node_file (str) – node feature and label file
edge_file (str) – edge list file
verbose (int, optional) – output verbose level
- property node_feature_dim#
Dimension of node features.
- property num_edge#
Number of edges.
- property num_node#
Number of nodes.
ReactionDataset#
- class ReactionDataset(*args, **kwds)[source]#
Chemical reaction dataset.
Each sample contains two molecule graphs, and any number of prediction targets.
- load_smiles(smiles_list, targets, transform=None, verbose=0, atom_feature='default', bond_feature='default', mol_feature=None, with_hydrogen=False, kekulize=False, node_feature=None, edge_feature=None, graph_feature=None)[source]#
Load the dataset from SMILES and targets.
- Parameters
smiles_list (list of str) – SMILES strings
targets (dict of list) – prediction targets
transform (Callable, optional) – data transformation function
verbose (int, optional) – output verbose level
atom_feature (str or list of str, optional) – atom features to extract
bond_feature (str or list of str, optional) – bond features to extract
mol_feature (str or list of str, optional) – molecule features to extract
with_hydrogen (bool, optional) – store hydrogens in the molecule graph. By default, hydrogens are dropped
kekulize (bool, optional) – convert aromatic bonds to single/double bonds. Note this only affects the relation in
edge_list. Forbond_type, aromatic bonds are always stored explicitly. By default, aromatic bonds are stored.node_feature (str or list of str, optional) – deprecated alias of
atom_featureedge_feature (str or list of str, optional) – deprecated alias of
bond_featuregraph_feature (str or list of str, optional) – deprecated alias of
mol_feature
- property atom_types#
All atom types.
- property bond_types#
All bond types.
- property edge_feature_dim#
Dimension of edge features.
- property node_feature_dim#
Dimension of node features.
- property num_atom_type#
Number of different atom types.
- property num_bond_type#
Number of different bond types.
SemiSupervised#
Data Processing#
DataLoader#
- class DataLoader(dataset, batch_size=1, shuffle=False, sampler=None, batch_sampler=None, num_workers=0, collate_fn=<function graph_collate>, **kwargs)[source]#
Extended data loader for batching graph structured data.
See torch.utils.data.DataLoader for more details.
- Parameters
dataset (Dataset) – dataset from which to load the data
batch_size (int, optional) – how many samples per batch to load
shuffle (bool, optional) – set to
Trueto have the data reshuffled at every epochsampler (Sampler, optional) – sampler that draws single sample from the dataset
batch_sampler (Sampler, optional) – sampler that draws a mini-batch of data from the dataset
num_workers (int, optional) – how many subprocesses to use for data loading
collate_fn (callable, optional) – merge a list of samples into a mini-batch
kwargs – keyword arguments for torch.utils.data.DataLoader
Dataset Split Methods#
- graph_collate(batch)[source]#
Convert any list of same nested container into a container of tensors.
For instances of
data.Graph, they are collated bydata.Graph.pack.- Parameters
batch (list) – list of samples with the same nested container
- ordered_scaffold_split(dataset, lengths, chirality=True)[source]#
Split a dataset into new datasets with non-overlapping scaffolds and sorted w.r.t. number of each scaffold.
- Parameters
dataset (Dataset) – dataset to split
lengths (list of int) – expected length for each split. Note the results may be different in length due to rounding.
Feature Functions#
Atom Features#
- atom_default(atom)[source]#
Default atom feature.
- Features:
GetSymbol(): one-hot embedding for the atomic symbol
GetChiralTag(): one-hot embedding for atomic chiral tag
GetTotalDegree(): one-hot embedding for the degree of the atom in the molecule including Hs
GetFormalCharge(): one-hot embedding for the number of formal charges in the molecule
GetTotalNumHs(): one-hot embedding for the total number of Hs (explicit and implicit) on the atom
GetNumRadicalElectrons(): one-hot embedding for the number of radical electrons on the atom
GetHybridization(): one-hot embedding for the atom’s hybridization
GetIsAromatic(): whether the atom is aromatic
IsInRing(): whether the atom is in a ring
- atom_symbol(atom)[source]#
Symbol atom feature.
- Features:
GetSymbol(): one-hot embedding for the atomic symbol
- atom_position(atom)[source]#
Atom position in the molecular conformation. Return 3D position if available, otherwise 2D position is returned.
Note it takes much time to compute the conformation for large molecules.
- atom_property_prediction(atom)[source]#
Property prediction atom feature.
- Features:
GetSymbol(): one-hot embedding for the atomic symbol
GetDegree(): one-hot embedding for the degree of the atom in the molecule
GetTotalNumHs(): one-hot embedding for the total number of Hs (explicit and implicit) on the atom
GetTotalValence(): one-hot embedding for the total valence (explicit + implicit) of the atom
GetFormalCharge(): one-hot embedding for the number of formal charges in the molecule
GetIsAromatic(): whether the atom is aromatic
- atom_explicit_property_prediction(atom)[source]#
Explicit property prediction atom feature.
- Features:
GetSymbol(): one-hot embedding for the atomic symbol
GetDegree(): one-hot embedding for the degree of the atom in the molecule
GetTotalValence(): one-hot embedding for the total valence (explicit + implicit) of the atom
GetFormalCharge(): one-hot embedding for the number of formal charges in the molecule
GetIsAromatic(): whether the atom is aromatic
- atom_pretrain(atom)[source]#
Atom feature for pretraining.
- Features:
GetSymbol(): one-hot embedding for the atomic symbol
GetChiralTag(): one-hot embedding for atomic chiral tag
- atom_center_identification(atom)[source]#
Reaction center identification atom feature.
- Features:
GetSymbol(): one-hot embedding for the atomic symbol
GetTotalNumHs(): one-hot embedding for the total number of Hs (explicit and implicit) on the atom
GetTotalDegree(): one-hot embedding for the degree of the atom in the molecule including Hs
GetTotalValence(): one-hot embedding for the total valence (explicit + implicit) of the atom
GetIsAromatic(): whether the atom is aromatic
IsInRing(): whether the atom is in a ring
- atom_synthon_completion(atom)[source]#
Synthon completion atom feature.
- Features:
GetSymbol(): one-hot embedding for the atomic symbol
GetTotalNumHs(): one-hot embedding for the total number of Hs (explicit and implicit) on the atom
GetTotalDegree(): one-hot embedding for the degree of the atom in the molecule including Hs
IsInRing(): whether the atom is in a ring
IsInRingSize(3, 4, 5, 6): whether the atom is in a ring of a particular size
IsInRing() and not IsInRingSize(3, 4, 5, 6): whether the atom is in a ring and not in a ring of 3, 4, 5, 6
Bond Features#
- bond_default(bond)[source]#
Default bond feature.
- Features:
GetBondType(): one-hot embedding for the type of the bond
GetBondDir(): one-hot embedding for the direction of the bond
GetStereo(): one-hot embedding for the stereo configuration of the bond
GetIsConjugated(): whether the bond is considered to be conjugated
- bond_length(bond)[source]#
Bond length in the molecular conformation.
Note it takes much time to compute the conformation for large molecules.
Residue Features#
Molecule Features#
- ExtendedConnectivityFingerprint(mol, radius=2, length=1024)[source]#
Extended Connectivity Fingerprint molecule feature.
- Features:
GetMorganFingerprintAsBitVect(): a Morgan fingerprint for a molecule as a bit vector
- ECFP()#
alias of
torchdrug.data.feature.ExtendedConnectivityFingerprint
Element Constants#
Element constants are provided for convenient manipulation of atom types. The atomic numbers can be accessed by uppercased element names at the root of the package. For example, we can get the carbon scaffold of a molecule with the following code.
import torchdrug as td
from torchdrug import data
smiles = "CC1=C(C=C(C=C1[N+](=O)[O-])[N+](=O)[O-])[N+](=O)[O-]"
mol = data.Molecule.from_smiles(smiles)
scaffold = mol.subgraph(mol.atom_type == td.CARBON)
mol.visualize()
scaffold.visualize()
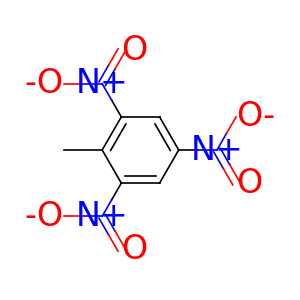
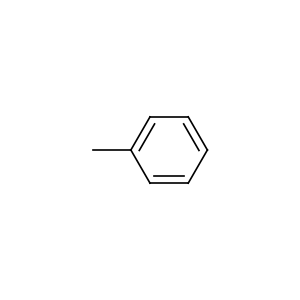
There are also 2 constant arrays that map atomic numbers to element names.
td.ATOM_NAME[i] returns the full name, while td.ATOM_SYMBOL[i] returns the
abbreviated chemical symbol for atomic number i.
For a full list of elements, please refer to the perodic table.